Efficient and Effective Delivery of AAV Therapies to Patients via Candidate Screening and Feasibility Studies
As drug developers embark on the early leg of their adeno-associated virus (AAV) therapy research and development journeys, drug candidate screening and feasibility studies help achieve preclinical and Phase 1 goals while minimizing cost and time spent. Early-phase assessment studies aim to create simple, modular paths to current good manufacturing practice (cGMP) to minimize risk and streamline drug substance and drug product manufacturing. When assessing potential manufacturing partners, consider a contract development and manufacturing organization (CDMO) that offers candidate screening and feasibility studies to optimize your process and expedite your path to cGMP.
Navigate The Current AAV Manufacturing Climate
The last two years have proved challenging for the viral gene therapy community. Drug developers report being up against the following challenges and limitations:
- A lack of adequate funding to commit to a full program.
- More discrete funding packages that require milestones to be hit before raising additional funds.
- Many inexperienced CDMO options in a market saturated with new development and manufacturing capacity.
As a result of these factors, the need to outsource end-to-end construct design through Phase 1 to larger CDMOs is growing. Therapy developers are looking to extract as much product out of research grade development activities as possible, leverage research use only (RUO) supply, and ensure that construct design is right the first time, such that you have sufficient understanding to determine the commercial viability of your drug in a cost-efficient manner. To keep the R&D, preclinical, and clinical processes as continuous as possible, it is important to consider key bioprocess opportunities during early-phase construct design through to process development and cGMP manufacture.
Your CDMO should understand the challenges that investigators face when it comes to raising funding, and in turn, offer solutions, such as candidate screening and feasibility studies to provide material that enables generation of clinical data to support the next round of funding.
Assess The Impact of Candidate Screening and Feasibility Studies
I. Candidate Screening
In the codon and promoter optimization world, there could be four to 15 candidates with different promoters, codon optimizations, and gene of interest strategies regarding sizing and serotype design in a tissue that has multiple AAV serotypes. FUJIFILM Diosynth Biotechnologies (FDB) has built a solid foundation from candidate screening and feasibility through to phase-appropriate manufacture.
Candidate screening requires a minimal amount of bioprocessing, generally a shake flask or small stirred tank bioreactor’s worth. It allows screening of multiple different candidates simultaneously which are run through a common downstream process (Figure 1). Previously, less readily scalable methods like ultracentrifugation or precipitation followed by ultracentrifugation were commonly used. At FDB, we moved away from these to identify a more manufacturing ready process to rapidly evaluate as many different constructs as possible.
The number of constructs we can evaluate downstream depends on purity and serotypes required. Downstream raw materials such as chromatography resins that are largely serotype agnostic are run through a representative process to provide material that eventually will be consistent with what is needed for IND-enabling, toxicology, and early-phase studies. We also conduct a pared down analytical panel that is focused on critical quality attributes common to AAV, including titer using digital droplet polymerase chain reaction (ddPCR) with inverted terminal repeats (ITR) or gene of interest (GOI)-specific primer probe sets, and full to empty ratio using mass photometry.
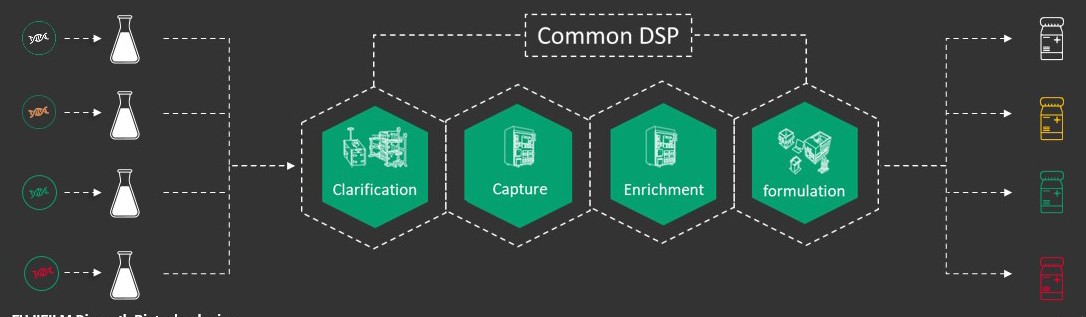
Figure 1: Candidate screening workflow
II. Feasibility Studies
Once construct design is complete and we have narrowed down the most promising two or three AAV constructs from a manufacturing and/or clinical efficacy standpoint, we scale up and run a more optimized process with 10 L bioreactors. The feasibility study has a similar core process to candidate screening; it should be noted that establishing continuity between the construct design, top three candidates, and the feasibility study is important. The formulation is critical, and it is difficult to formulate with only a few liters of material. Feasibility studies include scale-up, additional material for your preclinical studies, biodistribution analysis, and dose range finding prior to toxicology design. They also provide more expansive product quality and residual analytics, including genome titer, full to empty ratio analysis, capsid titers, residual host cell DNA, and host cell protein. These are essential to ensuring process performance and scalability.
If a developer and CDMO identify critical quality attributes that may be challenging to achieve, it is beneficial to start with a feasibility study, perform a platform compatibility assessment to determine if further development is needed, and adjust process development (Figure 2). As we push AAV concentrations into and above 5 x 1,014 vg/mL, residuals become a greater concern. By the time you move into the demonstration run, clear success criteria have been identified that must be achieved to supply material for a toxicology study that precedes cGMP manufacture. These feasibility assessments make the middle three steps – platform comparability assessment, process development, and demonstration run – as efficient as possible from a time and cost of goods perspective.
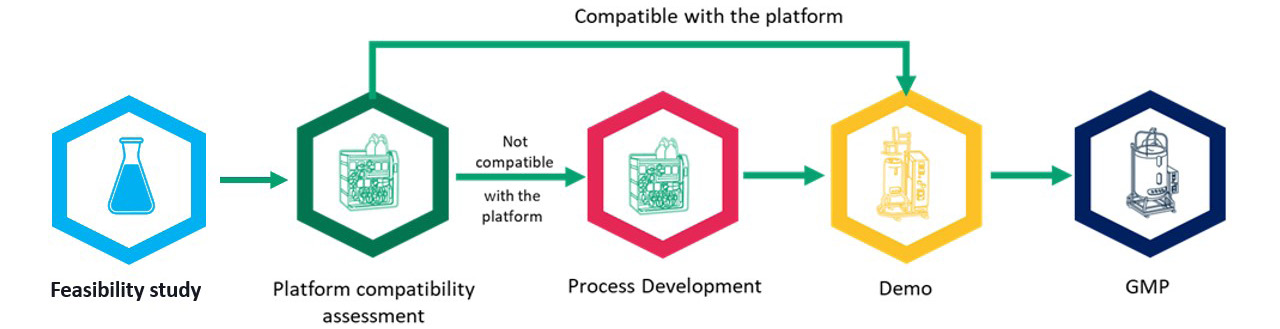
Figure 2: The benefits of feasibility studies
Examine an AAV Feasibility Case Study
A client contracted FDB to perform a feasibility study on their leading AAV9 candidate. This study entailed running a series of 10 L bioreactors to investigate transfection parameters. All the bioreactor runs achieved the customer’s target threshold of >1 x 1,011 vg/mL at harvest. For most clinical indications, this titer is typically an inflection point between manufacturable and non-manufacturable, depending upon dose and concentration. The 10 L reactors were processed through FDB’s standard downstream process for AAV9, resulting in a process yield of around 40%, with the resultant purified material possessing 80% full capsids. Having achieved the client‘s goals, the process was scaled up to provide material for toxicology studies. The toxicology supply required two synchronized 200 L bioreactors that were combined into a single downstream purification. These runs were successfully executed, achieving comparable titers, yields, and % full capsids. Our results demonstrated that running these processes at a small scale with a representative bioprocess can provide biodistribution and dose range finding material while maintaining consistency with toxicology and cGMP.
Start a CDMO Relationship on the Right Foot
At the outset of a partnership, FDB will provide your team with a screening request form that gathers relevant information about the clinical indication, including concentration, host cell residuals, purity analysis, titer, and full to empty ratio. This information enables the process development team to build a scale-appropriate process that delivers representative material in the agreed-up-on timelines. Whether you’re looking to start with multi-candidate screening or at a later stage, there are a variety of entry points at FDB (Figure 3).
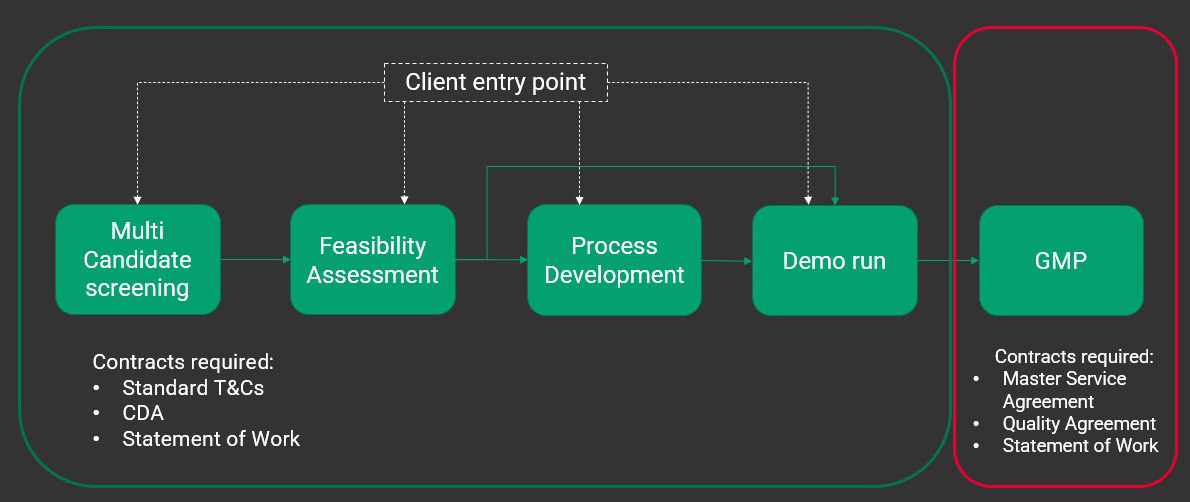
Figure 3: Research grade entry points vs. GMP entry points Entry points contained within the green box are all non GMP in nature. These contracts require standard terms and conditions, a confidentiality agreement, and a statement of work that outlines what needs to be delivered to the customer and what the customer should expect. GMP work requires additional contracts including a master service agreement and quality agreement in addition to those required for non-GMP work.
Consider The Benefits of a Modular Approach
FDB has developed several flexible modular offerings that offer clients lower risks and costs (Figure 4). Each stage has defined criteria based on the scope of work and key deliverables. These approaches allow for rapid generation of R&D material for preclinical work, a streamlined process to IND, and an efficient shift from preclinical to clinical without significant process changes, allowing the scope of a project to grow with a client’s needs. Our candidate screening and feasibility assessments are just the beginning of a partnership.
Upon completion of those early studies, programs typically progress through small-scale process development and optimization. From there, we ensure that toxicology material is provided in a timely manner and meets agreed critical quality attribute criteria. FDB has the capacity to support our clients throughout the full life cycle of their product, from preclinical supply through to commercial manufacture. To meet your goals, we collaborate to identify the most suitable modules and the necessary deliverables. We aim to deliver the right product at the right specification so that it can be administered to patients with maximum clinical efficacy.
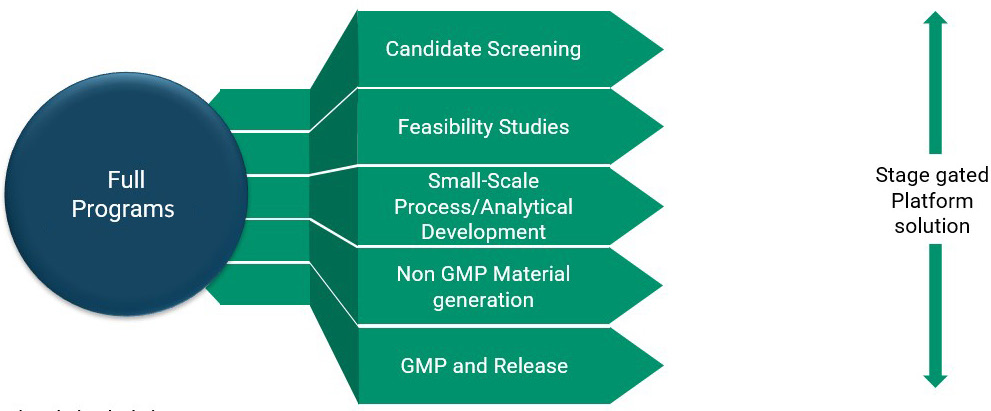
Figure 4: Modular approaches to full programs
Choose What Works For You
To manufacture your AAV gene therapy with high quality and efficiency, work with an experienced AAV manufacturer that prioritizes innovative and effective technologies to meet clinical challenges while employing experienced teams that understand how to advance your product from preclinical development to commercialization. At FDB, we stay ahead of the learning curve so our clients can deliver next-generation therapeutics to patients via seamless, consistent, and predictive modules. If you are curious about how candidate screening and feasibility studies could help you, view the recent webinar hosted by FDB on the impact of candidate and feasibility studies for AAV therapeutics or contact us directly here.
About the Author
Ian Goodwin is Director of CMC Program Design in Advanced Therapies covering Cell and Gene Therapy services. He has been with the company for six years, having also served as Director of Upstream Process Development and in scientific roles within the Process Development group. Ian is currently responsible for providing strategic CMC guidance, technical design, and project scope development support to prospective clients engaged in CDMO selection for viral and non-viral cell & gene therapy products. Ian’s previous experience includes 10+ years working on process development and manufacturing strategies for stem cell, regenerative medicine, and gene therapy viral vectors, specifically adeno-associated virus (AAV) and lentivirus vectors (LV) in academic, start-up, and CDMO environments. Ian holds a Master’s Degree in Microbiology and Immunology from Drexel University College of Medicine and a B.Sc. in Microbiology from the University of Pittsburgh.
About FDB
FDB is a global CDMO operating in Europe and North America with integrated platforms to meet client demands and deliver medicines to patients faster. FDB has capabilities not only with drug substance manufacturing but also with finished goods manufacturing on both sides of the Atlantic, allowing a customer to work with a single provider for their end-to-end manufacturing needs. To learn more about their CDMO offerings, contact the FDB team.