Effectively Enabling Rare Diseases to Reach Patients
By Dr. Rebecca Abram, Director, Strategic Technical Marketing, and Dr. Joanna Norman, Director of PD Operations, Viral Gene Therapy Operations, U.K.
Source: FUJIFILM Diosynth Biotechnologies
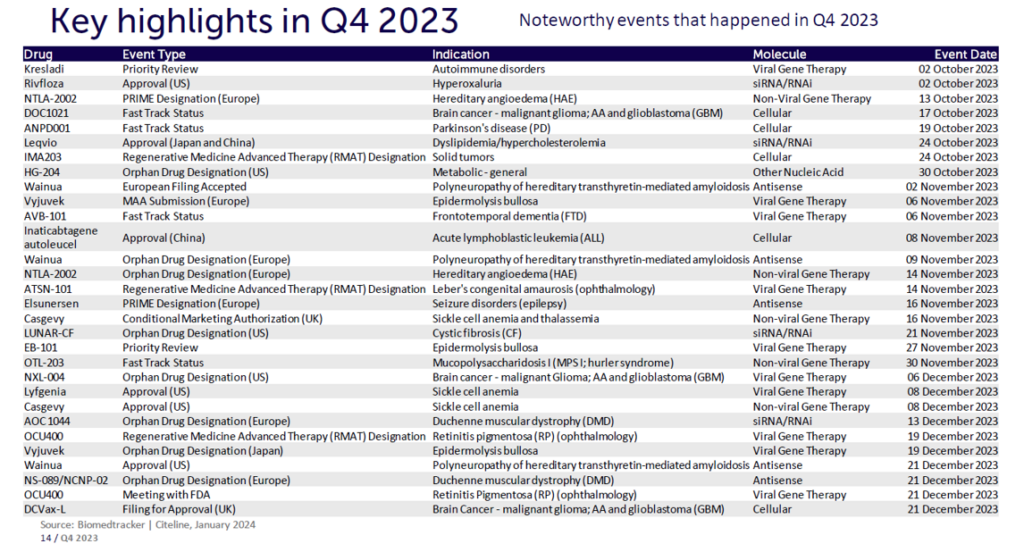
Rare diseases continue to represent a large unmet medical need globally. Despite their designation, the 7,000 known rare diseases affect over 400 million people worldwide. There is growing hope, though, for patients and their families. In Figure 1, the five recent rare disease approvals across the cell, gene, and RNA therapy landscape are indicated with a green dot. With rare disease therapies now in development by 323 companies worldwide and an ever-growing number of fast-track approvals and orphan designations, the future is primed to offer significantly more treatment options for rare disease patients.
Enter the Crowded Gene Therapy Landscape
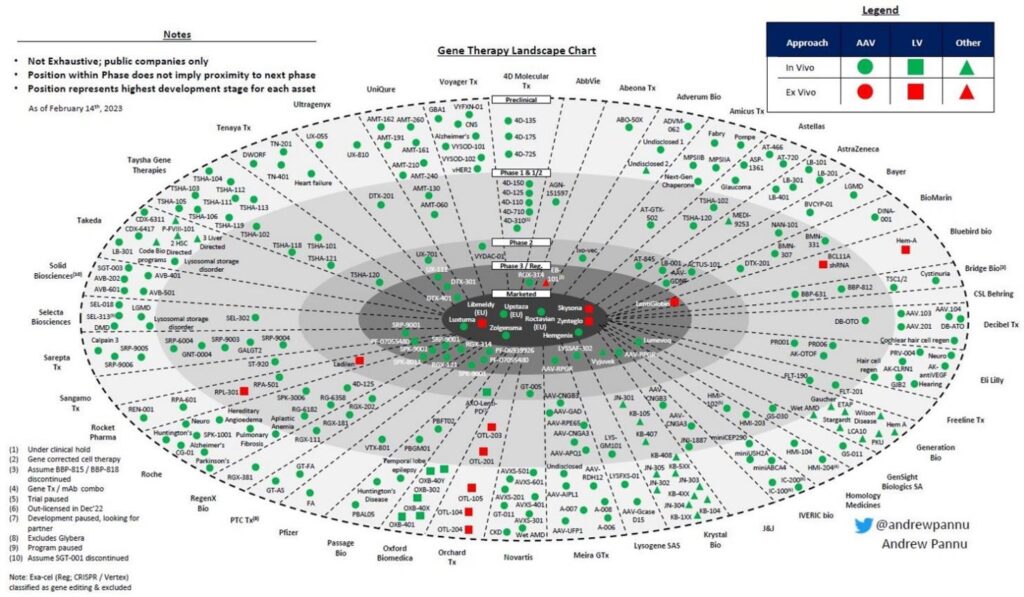
Despite the busy, early-phase gene therapy landscape, only eight therapies are on the market (Figure 2) and
patients remain in need of innovative treatments. However, taking rare disease therapies to clinic is challenging. Developers need a robust understanding of their readiness for scaleup and insight into how to manage timeline, budget, resources, and low patient numbers to ensure return on investment (ROI). Since these are emergent technologies, sponsors are often dealing with minimal standardization and guidance from the field as well as difficulty defining release specifications due to low batch numbers. Across the space, there is a need to better understand critical quality attributes (CQAs) and their impact.
Beyond that, developers are up against potential clinical holds. According to the National Institute of Health (NIH), though cell and gene therapies only account for 2% of clinical trials, they represent 40% of clinical holds over the last five years.1 Of these holds, 22% were a response to the FDA’s request for more chemistry, manufacturing, and controls (CMC) information.1 They lasted 100 days on average, which can be detrimental for patients seeking treatment as well as drug developers forced to reset their pipeline.1 Peter Marks, the FDA’s Director of the Center for Biologics Evaluation and Research, has spoken on the FDA’s commitment to expand cell and gene therapy approvals, noting that increased holds are often the result of inexperienced sponsors, and emphasizing the importance of early, frequent communication with the
FDA.2
Enlist the Help of an Expert
To help counteract a lack of previous experience, sponsors should consider partnering with a CDMO that offers ample development, manufacturing, engineering, quality, and regulatory expertise. Look for the following indicators:
- Skilled experts that leverage high-throughput analytical
processing technologies - Integrated capabilities that reduce the need for
too many suppliers - Flexibility and capacity to manufacture at multiple
scales - Regulatory experts that assist with developing
submission packages
While choosing an experienced CDMO is important, so too is partnering at the beginning of development to set your goals, cost, and timeline. By partnering early, clients establish formulation and process parameters so that if something should change, they understand the impact on the process. Furthermore, they can collaboratively design a risk-based CMC plan to share with regulatory authorities as soon as possible.
Recognize the Hallmarks of
Experience
At FUJIFILM Diosynth Biotechnologies (FDB), we offer our clients end-to-end solutions to accommodate their development of rare disease therapies. We are cognizant of potential challenges and have worked to identify solutions to facilitate a smooth progression through CMC milestones. FDB has developed processes for adeno-associated virus (AAV), adenovirus, lentivirus, herpes simplex virus (HSV), and pox viruses; accommodated Phase 1 to Phase 3 CMC submissions; and is currently guiding a partner through process characterization and validation to support commercialization.
Our experience encompasses both suspension and adherent production platforms, and we implement the appropriate propagation strategy based on a client’s needs. For AAV programs, we have experience with triple transfection, sf9/baculovirus expression, HSVmediated production, and capabilities using producer cell lines. For lentivirus, we have leveraged the quad-transfection system and demonstrated infection strategies for adenovirus, HSV, and pox viruses. For AAV products, our scaling capabilities can go up to 500 L in suspension and can implement up to 200 L suspension for adenovirus and lentivirus therapies. We have also implemented manufacturing strategies for adherent products, including microcarriers and fixed
bed bioreactors.
Our streamlined platform approaches and associated analytics help improve affordability, minimize process development, and increase productivity. Many critical reagents are provided in-house, including cell lines and transfection plasmids, allowing us to start projects without external sourcing. We strive to understand CQAs beyond platform assays and develop and leverage therapy-specific potency assays. For rare diseases, it is critical to progress from development to clinic rapidly. FDB’s programs are structured with regulatory submissions at the forefront, and CMC considerations are built into the project. Our experience scaling up processes and working with regulatory agencies allows us to support a range of partners.
Choose the Entry Point that Works
for You
No two developers arrive at a CDMO with the same project needs. To accommodate this, FDB has
designed discrete packages to offer clients flexibility to suit their budgetary and timeline goals. We offer multiple entry points: multi-candidate screening, feasibility assessments, process development, and demo run to scaleup. Opting to conduct candidate screening and/or feasibility studies helps ensure you have a representative process from the outset and allows you to solve potential challenges prior to scaleup. It also minimizes bridging studies caused by significant process changes.
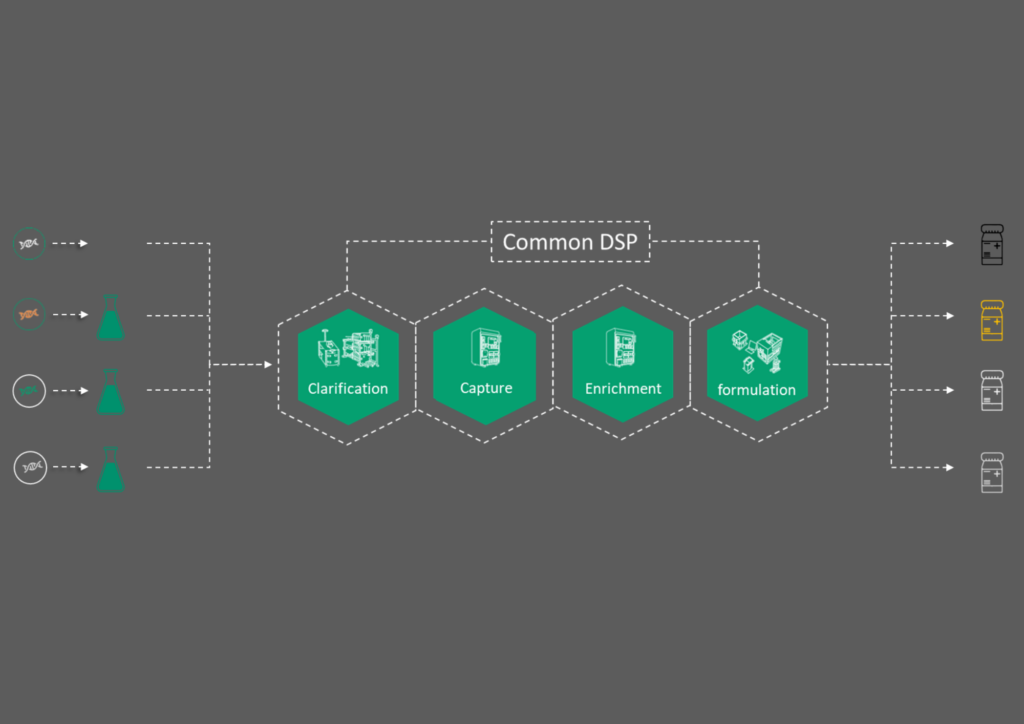
Candidate screenings assess which constructs are most productive and which show indications of the required CQAs to progress to feasibility (Figure 3). We perform candidate screening on up to six constructs, followed by 6 L lab scale runs to generate material for the selected candidate. Our team runs through a standard upstream process with optimized conditions for plasmid ratio and transfection, and then processes the material through the downstream using predefined conditions. The material can be formulated into a buffer of our client’s choosing, and we offer a customizable analytical panel. We generally conduct digital droplet polymerase chain reaction (ddPCR) to look at genome content and enzyme-linked immunosorbent assay (ELISA) to assess titer.

Our feasibility study is a lab scale run to test the platform or generate material once a lead candidate has been chosen. We run a 10 L bioreactor process through the workflow shown in Figure 4 followed by a reduced testing panel that includes genome content (ddPCR), capsid titer (ELISA), and empty versus full by analytical ultracentrifugation (AUC). We assess residuals for host cell protein (HCP) and DNA, as well as aggregation.
Overall, the process is fast, flexible, and straightforward, allowing clients an adaptable scope to reach their goals and maintain their budget. Candidate screening and feasibility studies are low cost, and material generation time is relatively short. Following feasibility, we work with customers to review data and identify areas of improvement to ensure that CQA targets are met. From there, we conduct a demo run at the scale if required before implementing the process at manufacturing scale. Our established early-stage processes offer a smooth, streamlined path to good manufacturing practice (GMP).
Explore a Standard AAV Process
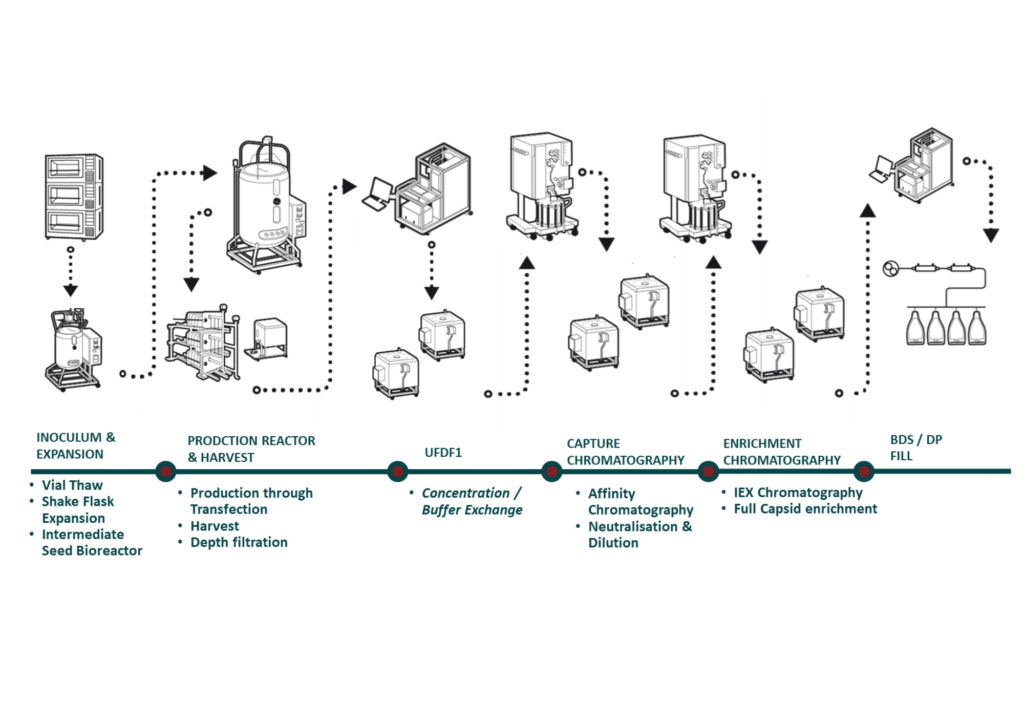
Figure 5 details a typical AAV process at scale, starting in shake flask, moving through an intermediate seed reactor, followed by the production reactor. Our equipment is aligned to the smaller scales, which minimizes scaling issues. The AAV platform package comes with established analytical methods, and we use phase-appropriate method validation tailored to the needs of the program. FDB determines genomic titer by ddPCR / qPCR and capsid titer by ELISA and high-performance liquid chromatography (HPLC). We utilize ITR elements for genomic titer initially, and then transition to gene of interest (GOI) titer for later stages in the program. The residuals are already established and well understood, which is standard for our AAV platform programs. The team uses multiplexing strategies to generate more data in a single run to keep costs low and reduce timelines. In terms of characterization, small tweaks to pre-established methods may be required depending on the product. Our experts can develop product specific potency assays or tech transfer customers into FDB with ease. Mass spectrometry is utilized initially to gain a snapshot into the empty versus full ratio during development and confirmed through AUC.
Overall, FDB’s AAV platform demonstrates consistent productivity from 10 to 200 L and high yields throughout the process. In a typical product profile, we achieve an empty versus full ratio of 20 to 30%. We meet regulatory requirements for HCP in host cell DNA clearance across the process, maintain a high purity profile, and typically see a two to threefold enrichment of empty versus full across the process achieving an
overall process yield of roughly 30%.
Prioritize Quality and Efficiency
Bringing rare disease therapy to patients is vital but challenging. The right CDMO partner will meet you where you are and guide you through your process development and scale up to ensure you are putting your best foot forward for regulatory submissions. Though platforms are not one-size-fits-all, they aid sponsors in taking products to clinic in the most cost-effective way.
If you’re interested in learning more, view the full webinar that was hosted by FDB on rare diseases day in February 2024 or contact us directly for more information.
References
1. Wills, C. A., Drago, D., & Pietrusko, R. G. (2023). Clinical holds for cell and gene therapy trials: Risks, impact, and lessons learned. Molecular therapy. Methods & clinical development, 31, 101125.
2. Brennan, Z. (2023, February 13). Operation warp speed for rare diseases: CBER leader says pilot is coming soon. Endpoints News. https://endpts.com/operation-warp-speed-for-rare-diseases-cber-leader-says-pilot-is-coming-soon/
About the Authors

Rebecca Abram holds the position of Director, Strategic Technical Marketing at FUJIFILM Diosynth Biotechnologies. She has global responsibility for leading technical marketing activities for all service offerings in alignment with our Commercial and Business Strategic Groups. Rebecca joined FUJIFILM Diosynth Biotechnologies in Billingham, UK, in the Analytical Development department in 2019 and led the analytical component for over 70+ early and late phase customer programs. Rebecca completed post-doctoral research at Durham University (UK) in collaboration with Avecia into the plasticity and wound healing capabilities of adult hair follicle stem cells before and after bioreactor expansion. She attended The University of Sheffield (UK) where she received her BSc in Chemistry and subsequently completed her doctoral research into antiinflammatory
peptides.

Joanna Norman is Director of Viral Gene Therapies, Process Development Operations within FUJIFILM Diosynth
Biotechnologies. She oversees upstream, downstream and analytical operations at the Darlington (UK) site. Joanna joined FUJIFILM Diosynth Biotechnologies in 2012 as a Principal Scientist in Downstream Operations at the Billingham location. She has held a number of delivery and technical roles at the company’s sites, spending 2.5 years as a Program Manager delivering early and late phase VGT programs in College Station, Texas. Joanna attended Newcastle University (UK) where she earned her BSc in Medical Microbiology and Immunology and her PhD in Molecular Biology and Biochemistry.
About FUJIFILM Diosynth Biotechnologies
FUJIFILM Diosynth Biotechnologies, a subsidiary of FUJIFILM Corporation, is a world-leading contract development and manufacturing organization (CDMO) for the development and manufacture of biologics, advanced therapies, and vaccines. The company operates a global network with major
locations in the Unites States of America, the United Kingdom and Denmark, offering end-to-end services including drug substance, drug product, and finished goods services. It is also building a new manufacturing site in Holly Springs, North Carolina, USA, scheduled to be operational in 2025.
FUJIFILM Diosynth Biotechnologies has over thirty years of experience in developing and manufacturing drug substance of recombinant proteins, monoclonal antibodies, vaccines, among other large molecules, viral products and medical countermeasures expressed in a wide array of microbial,
mammalian, and host/virus systems. We have drug product filling capabilities to support both clinical and commercial demands. Our finished goods services, supported by more than 15 years of experience, can accommodate commercial products for more than 65 countries around the world. The
company offers a comprehensive list of services from cell line development using its proprietary pAVEway™ microbial and Apollo™X cell line system to process development, analytical development, clinical and FDA-approved commercial manufacturing. For more information, go to: www.fujifilmdiosynth.com.